
Descubra os sinais para se atentar às imunodeficiências na infância, assim como os exames mais importantes e quais são as deficiências de anticorpos mais frequentes.
Conforme relembramos no texto “Imunodeficiências na Infância – uma breve introdução“, as imunodeficiências primárias (IDP) são um grupo heterogêneo de doenças que afetam o desenvolvimento do sistema imune, sua função ou ambos. As manifestações podem ser:
- infecções recorrentes, graves e/ou por microorganismos de baixa patogenicidade;
- autoimunidade e inflamação;
- maior incidência de neoplasias.
As deficiências predominantes de anticorpos são as mais comuns, caracterizando pouco mais da metade dos casos, conforme o gráfico abaixo demonstra [1].
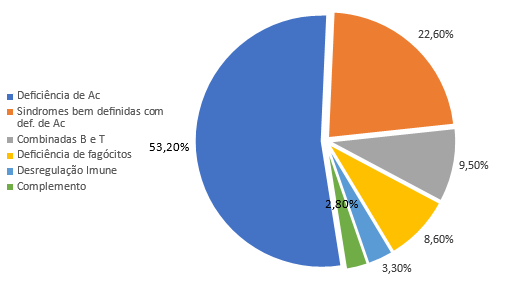
Assim, na prática pediátrica temos grandes chances de nos deparar com pacientes com IDP. Um grande exemplo disso é a deficiência de IgA — diversos estudos já apontavam para a grande prevalência da deficiência de IgA na população:
- 1:1.000 (Def. IgA — assintomáticos) [2]
- 1:50 (Def. IgA — Asma grave) [3]
Quando pensar em deficiências de anticorpos?
As deficiências de anticorpos se apresentam por infecções de repetição, geralmente por germes comuns da comunidade, com destaque para as bactérias encapsuladas (ex.: Streptococcus pneumoniae e Haemophilus influenza tipo B). Manifestações comuns são otites, sinusites e pneumonias. Nas crianças, podemos observar também baixo peso, déficit de crescimento, infecções recorrentes das vias respiratórias e do trato gastrointestinal, como diarreia crônica.
Deficiência de anticorpos – sinais em crianças:
- infecções de repetição (especialmente bactérias encapsuladas)
- otites
- sinusites
- pneumonias
- baixo peso
- déficit de crescimento
- infecções das vias respiratórias
- infecções do trato gastrointestinal
O início das manifestações geralmente é a partir dos 6 meses, quando os anticorpos maternos passados durante a gestação já foram depletados [4, 5]. Há um hiato imunológico que ocorre entre a perda dos anticorpos maternos e produção adequada de anticorpos pela criança, durante o qual ocorre um período fisiológico de hipogamaglobulinemia, geralmente entre os 3 e 6 meses de idade (veja na Figura 1 abaixo). Justamente nesse período, quando a criança está fisiologicamente menos protegida, é que grande parte das famílias matricula suas crianças em creches, o que favorece a ocorrência de infecções.
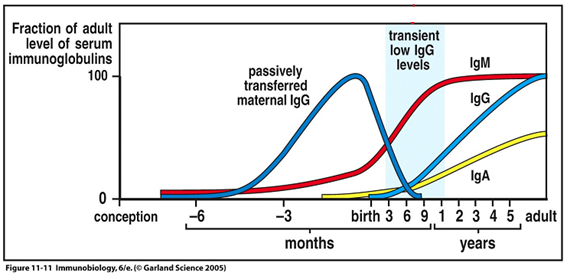
O prolongamento dessa fase durante a infância, com níveis baixos de IgG e, em alguns casos, também de IgA e IgM, é o que chamamos de hipogamaglobulinemia transitória da infância. Ressalta-se que esse termo nem sempre é bem adequado, pois em alguns casos a produção de anticorpos não irá ocorrer e a criança poderá apresentar algum outro diagnóstico, como imunodeficiência comum variável. Assim, o diagnóstico da hipogamaglobulinemia transitória da infância é retrospectivo [4].
Quais exames o pediatra pode realizar?
- Hemograma, com destaque para a série branca — leucopenia, linfopenia e neutropenia são sinais de alerta.
- Dosagem de Imunoglobulinas: IgG, IgA e IgM — lembrando que elas são variáveis de acordo com a idade. Alterações nos exames devem ser referenciadas a um profissional especializado.
Um especialista em imunologia pode solicitar exames adicionais, como a dosagem de subclasses de IgG, avaliação de respostas vacinais, respostas a antígenos polissacárides e subpopulações de linfócitos, entre outros, para melhor esclarecimento do quadro.
Imunodeficiência – diagnósticos diferenciais
Pacientes podem apresentar redução nos níveis de anticorpos secundários a outras condições, e devem ser pesquisadas de acordo com a história clínica de cada paciente.
| Causas de hipogamaglobulinemia associada a doenças | Causas de hipogamaglobulinemia associada a doenças |
| Doenças com perda proteica | Síndrome nefrótica Enteropatia perdedora de proteínas Grande queimado |
| Alteração de circulação linfática | Linfangiectasia intestinal Quilotórax Síndrome de Proteus |
| Doenças infecciosas | HIV na infância Infecções congênitas por rubéola, toxoplasmose, citomegalovírus, mononucleose |
| Aumento do catabolismo de imunoglobulinas | Distrofia miotônica Hiperesplenismo |
| Doenças de células B | Linfoma, mieloma multiplo, leucemia linfocítica crônica |
| Causas de hipogamaglobulinemia associadas a medicações | Causas de hipogamaglobulinemia associadas a medicações |
| Anticonvulsivantes | Carbamazepina Fenitoína Lamotrigina Valproato de sódio |
| Imunossupressores | Corticoides Ciclosporina Ciclofosfamina |
| Outros | Alguns imunobiológicos, captopril, clorpromazina, cloroquina |
Deficiências de anticorpos mais frequentes
Deficiência seletiva de IgA
É a mais comum das imunodeficiências. É definida pela presença de níveis baixos de IgA (menor que 7 mg/dL), com níveis normais de IgG e IgM, em pacientes maiores de 4 anos, após exclusão de causas secundárias. Pacientes que apresentam níveis de IgA acima de 7 mg/dL, porém abaixo de 2 desvios-padrões para a idade, são classificados como portadores de deficiência parcial de IgA. A grande maioria (85–90%) dos casos é de pacientes assintomáticos, porém uma parte pode apresentar infecções sinopulmonares e gastrointestinais de repetição. Esses pacientes apresentam maiores chances de desenvolver doenças autoimunes, como lúpus e artrite reumatoide. Existe maior prevalência de asma e atopia. [6]
Imunodeficiência Comum Variável (ICV)
Dentre as imunodeficiências graves, a ICV é a mais comum, com incidência estimada em 1:25.000 a 1:75.000. Consiste em um grupo heterogêneo de doenças caracterizado pela disfunção na produção de anticorpos. A IgG é baixa, associada também à redução de IgA ou IgM, com resposta inadequada à produção de anticorpos específicos e infecções recorrentes. [4]
Agamaglobulinemia
Na agamaglobulinemia, existe ausência ou redução muito acentuada da produção de anticorpos, além de ausência de células B. O início da ocorrência de infecções de repetição é precoce, entre 3 a 6 meses de vida, compatível com a queda dos anticorpos maternos de origem transplacentária. É mais comum em meninos, pois a forma mais frequente da doença, associada à mutação de uma tirosina quinase (tirosina quinase de Bruton), é ligada ao cromossomo X, sendo então chamada de XLA (agamaglobulinemia ligada ao X). [4]
Deficiência de subclasses de IgG
É a deficiência de uma ou mais subclasses de IgG (IgG1, IgG2, IgG3, IgG4), com níveis totais de IgG normais, podendo estar associada à deficiência de IgA. Seu diagnóstico pode ser difícil, visto que de 2 a 20% da população pode ter redução em alguma subclasse; além disso, os valores de referência variam com a idade. [4]
Deficiência específica de anticorpos antipolissacárides
É caracterizada pela falta de resposta a polissacarídeos, na presença de concentrações normais de IgG, IgA, IgM e IgE. Bactérias como o pneumococo e o Haemophilus influenza tipo B apresentam polissacarídeos nas suas cápsulas, sendo agentes frequentes de infecção. Para diagnóstico, além das dosagens de imunoglobulinas, utiliza-se a avaliação de resposta a vacinas polissacárides. Crianças abaixo de 2 anos apresentam, fisiologicamente, baixa resposta a esse tipo de estímulo. [4]
Tratamento das imunodeficiências humorais
O tratamento de imunodeficiências humorais depende da doença e de suas complicações, sendo realizado por especialistas.
Tratamentos específicos podem incluir:
- Reposição endovenosa ou subcutânea de imunoglobulina (quando indicado);
- Uso de antibiótico profilático (quando indicado);
- Tratamento das complicações;
- Vigilância sobre autoimunidades e neoplasias.
Resumindo…
- As imunodeficiências mais comuns são as de anticorpos (humorais).
- As principais manifestações das imunodeficiências humorais são infecções sinopulmonares, por bactérias encapsuladas; manifestações gastrointestinais também podem estar presentes;
- As dosagens de IgG, IgA e IgM, avaliadas de acordo com a faixa etária, podem fazer o diagnóstico de algumas das principais imunodeficiências. Se esses exames estiverem alterados, os se forem normais e ainda assim a suspeita de imunodeficiência persistir, o paciente deve ser encaminhado para avaliação especializada com um imunologista.
Referências
- Leiva LE, Zelazco M, Oleastro M, Carneiro-Sampaio M, Condino-Neto A, Costa-Carvalho BT, et al. Primary Immunodeficiency Diseases in Latin America: The Second Report of the LAGID Registry. J Clin Immunol. 2007 Jan 25;27(1):101–8.
- Carneiro-Sampaio MMS, Carbonare SB, Rozentraub RB et al. Frequency of selective IgA deficiency among Brazilian blood donors and healthy pregnant women. Allergol Immunopathol. 1987;17(4):213–6.
- Solé D, Zaha MM, Leser PG, Naspitz CK. Níveis de IgA na saliva de indivíduos normais e atópicos, determinados por anticorpos antiIgA secretora e antiIgA sérica. Rev Bras Alerg Imunol. 1987;10:120–5.
- Fried AJ, Bonilla FA. Pathogenesis, Diagnosis, and Management of Primary Antibody Deficiencies and Infections. Clin Microbiol Rev. 2009 Jul 1;22(3):396–414.
- Ballow M. Primary immunodeficiency disorders: Antibody deficiency. J Allergy Clin Immunol. 2002 Apr;109(4):581–91.
- Yel L. Selective IgA Deficiency. J Clin Immunol. 2010 Jan;30(1):10–6.




